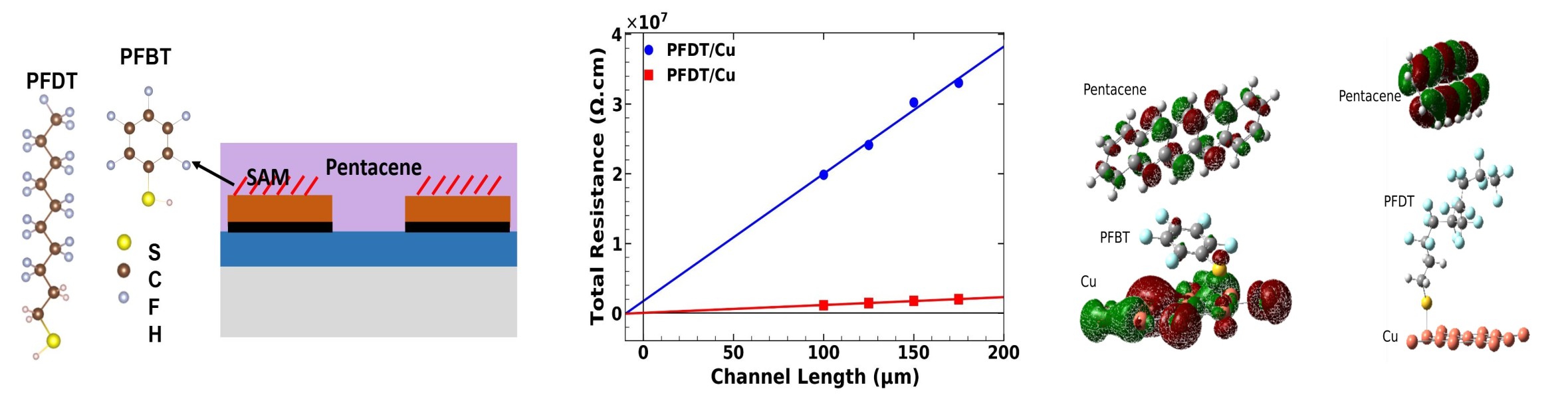
Contact interfaces in organic devices are highly resistive. Self Assembled Monolayers are commonly used as interlayers at these interfaces to reduce the resistance. However, there is still much ambiguity regarding the mechanism by which they facilitate the charge transfer at interface, so as to reduce the contact resistance. This has hindered their widespread use, as there are no guidelines driving molecular design for interlayers. The general understanding is that the polar molecules lead to a shift in work function of the contact material they are deposited on, thus modulating the energy level alignment at the interface with the organic semiconductor. This reduces the energy barrier to charge injection at the interface, and so facilitates charge transfer at the interface. However, there are exceptions to this. The shift in the work function does not always correlate with the reduction in contact resistance. I showed that SAM of PFBT and PFDT molecules reduced contact resistance by more than three orders in pentacene transistors with copper contacts. However, trends in contact resistance did not match with those in work function. I showed that the orbital delocalization at the interface plays a crucial role in governing the charge transport at the interface.
Based on this understanding, we developed an analytical function that correlated both the effects of work fucntion shift and interfacial orbital interactions with resulting device characteristics. The aim of the stuidy was to develop a computational model that could predict the performance of the SAM molecule in reducing contact resistance in organic devices. The interface stack was simulated and its properties computed by first principles. Considering electron transfer, the charge transfer integral J between lowest unoccupied molecular orbital of the organic semiconductor and the SAM functionalized contact was computed. Charge transfer intergal served as a measure of the extent of electronic coupling between the entities participating in charge transfer. We showed that the interface polarity, as represented by the partial charge on the semiconductor molecule (Qi>), is directly proportional to the work function of ligand functionalized electrodes. Thus a function combining J and Q comprehensively covers the microscopic factors governing charge transfer at the interface. We showed that the function is f= Q × log(J)
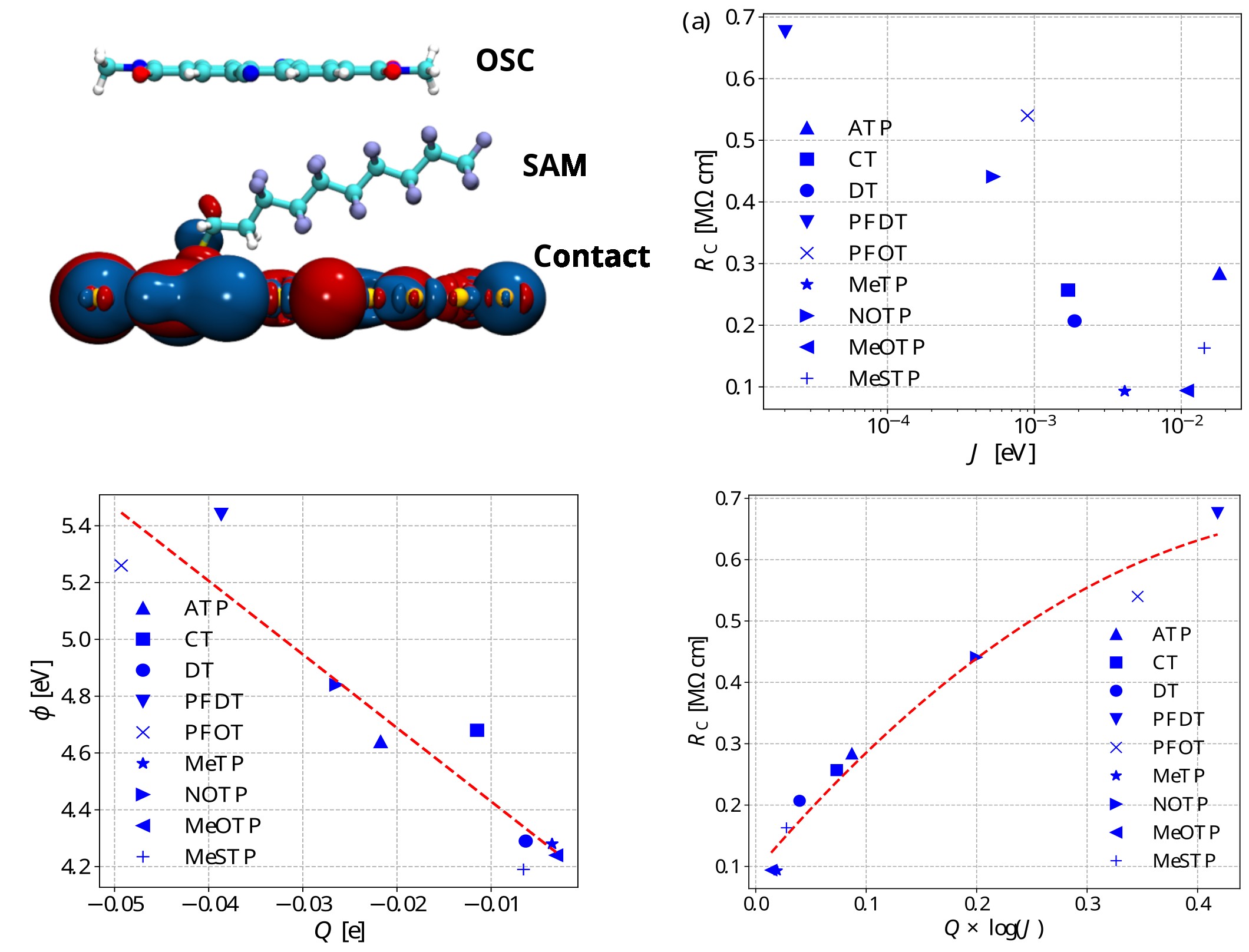
This establishes design guidelines for SAM molecules; a molecule ensuring higher value of J and lower value of Q would lead to the maximum reduction in contact resistance. The model also facilitates high throughput screening, by evaluating molecular structures directly.
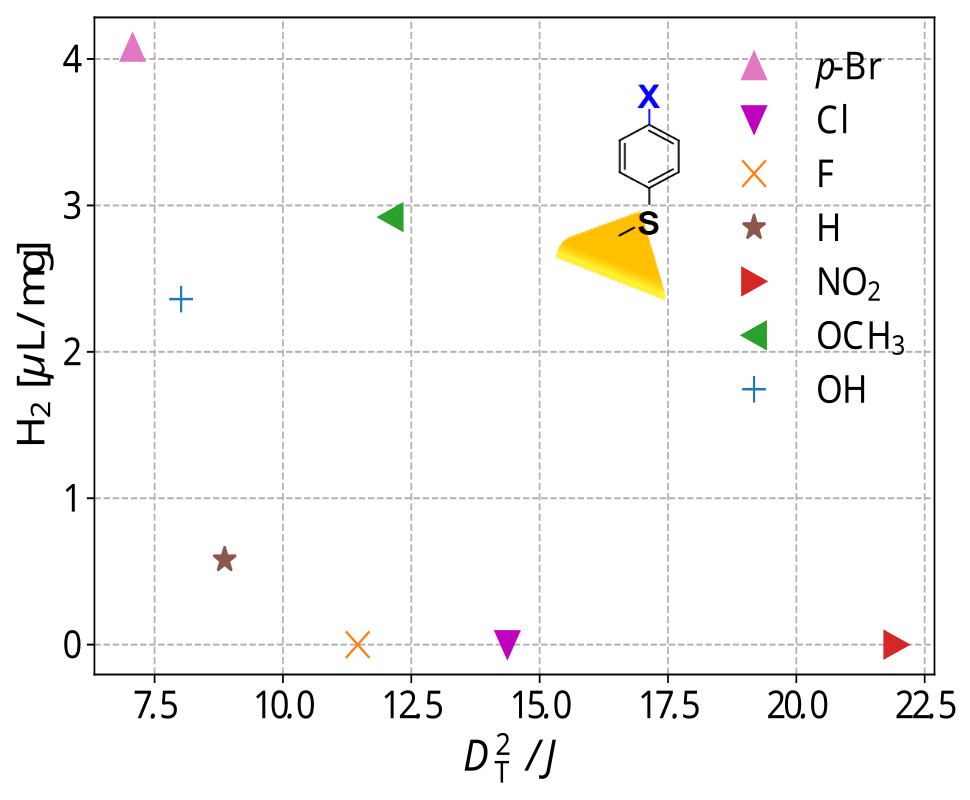
Further, this model was extended to design of ligands for photocatalysts. Thiolated ligands capped on Au nanoparticles have been found to be efficient photocatalysts for processes such as hydrogen production. The ligands facilitate charge transport from Au surface to reaction site, in a processd similar to that at contact interfaces in devices. Therefore, the facotrs determining catalytic activity are interface polarity (DT) and charge transfer integral here as well. We showed that a function of these quantities correctly predicted the trends in rate of hydrgen production for the photocatalyst with a given ligand.